Research
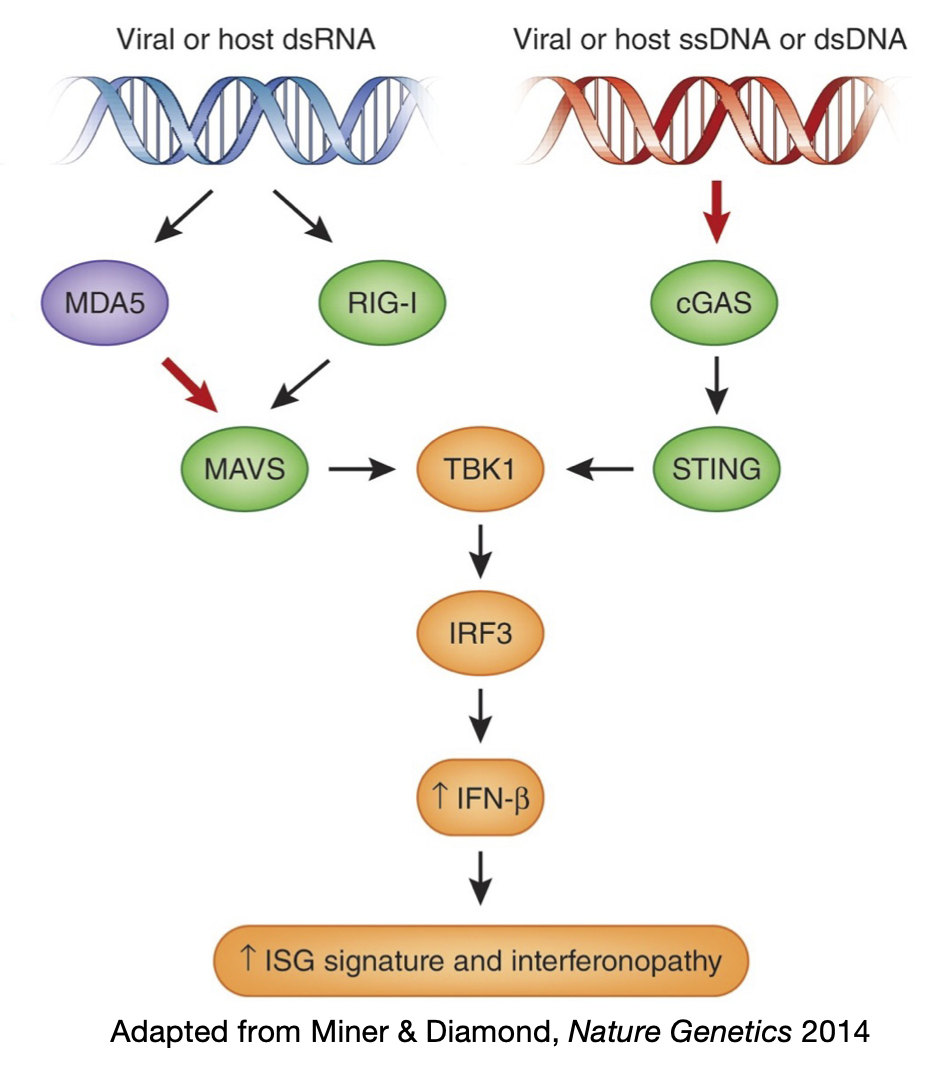
A hallmark of our immune system is the ability to discriminate between self and non-self molecules. To achieve this goal, cells utilize various sensors to recognize patterns frequently found in pathogens. These sensors are known as Pattern Recognition Receptors (PRRs). PRRs recognize DNA and RNA from microbial and viral pathogens. For example, in the cytosol, cGAS detects both viral and host DNA while RIG-I and MDA5 detect RNA. These sensors pass down the signals through different adapter proteins, which eventually merge to trigger the interferon (IFN) immune response, as the body’s first line of defense against viruses. Yet how cells avoid unwanted recognition of self molecules to prevent autoimmunity is poorly understood.
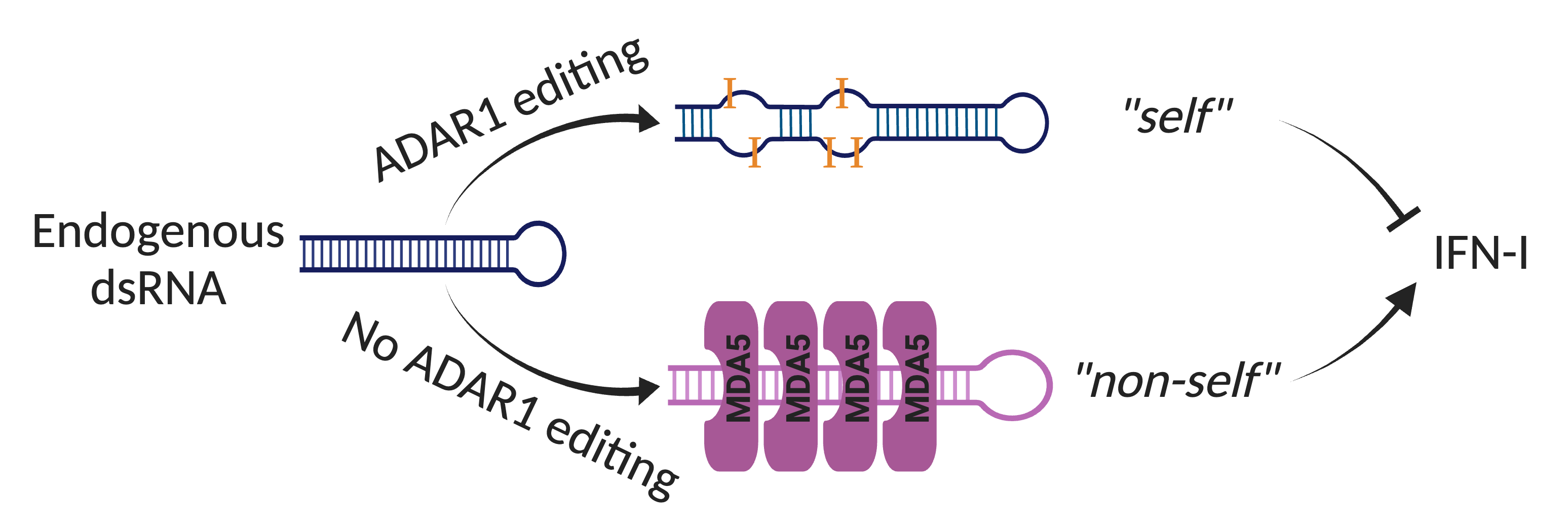
MDA5 is a cytosolic receptor that recognizes viral double-stranded RNAs (dsRNAs) to activate the antiviral IFN response. How does MDA5 avoid sensing self dsRNAs? The answer is Adenosine-to-Inosine (A-to-I) RNA editing catalyzed by ADAR that labels endogenous dsRNAs as self to evade MDA5 sensing. Mice deficient with ADAR1 editing are embryonic lethal, but can be rescued to full life span when MDA5 is removed. In humans, ADAR1 loss-of-function and MDA5 gain-of-function mutations lead to rare autoimmune diseases such as Aicardi-Gouteries Syndrome. While aberrant activation of MDA5 sensing is deleterious in healthy individuals, it can be beneficial in cancer immunotherapy. RNA editing occurs at millions of sites across human tissues (Tan, Li et al., Nature 2017), but it was unclear which dsRNAs, if not properly edited, are immunogenic, and what role the ADAR1-dsRNA-MDA5 axis plays in common diseases.
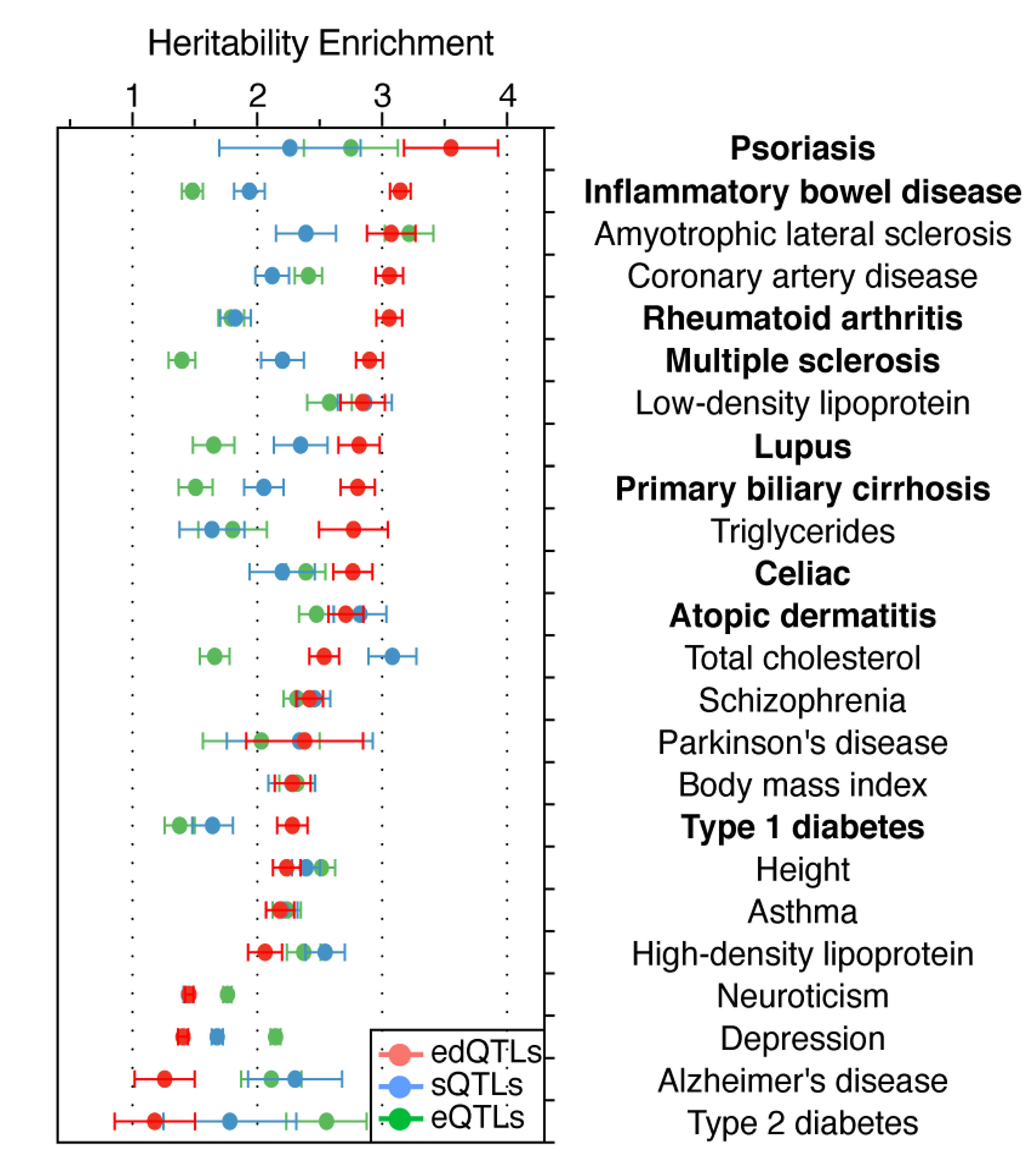
During Dr. Li’s postdoctoral training, he turned to human genetics for answers to these questions. He discovered that RNA editing quantitative trait loci (edQTLs) are strongly enriched in GWAS signals of common inflammatory diseases, even more than gene expression QTL and RNA splicing QTL. Furthermore, he showed that GWAS risk alleles, by each reducing editing level of their nearby immunogenic dsRNAs, can collectively elicit MDA5-dependent dsRNA sensing and subsequent chronic inflammation in a wide range of autoimmune and inflammatory diseases (Li et al., Nature 2022). These findings, based on well-established mechanism and human genetic evidence, show that insufficient RNA editing is a major risk factor underlying inflammatory diseases.
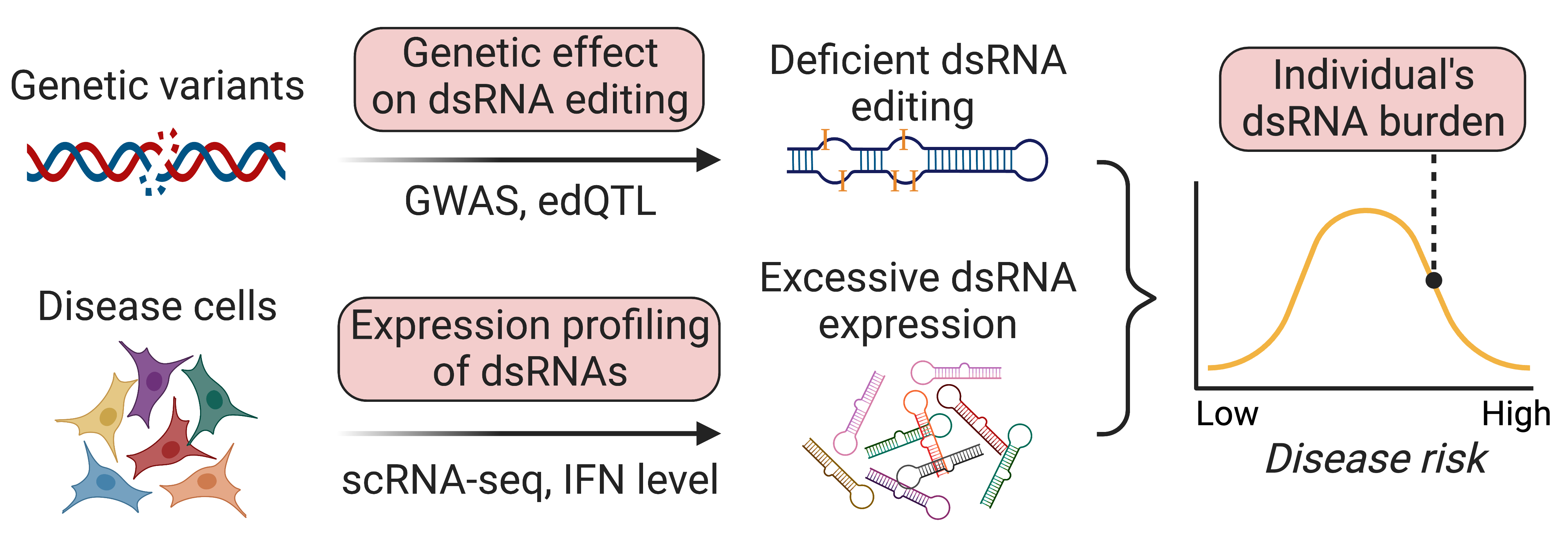
Our lab’s research goal is to advance basic understanding of dsRNA editing and sensing to develop better tools for diagnosis and treatment of inflammatory disease patients. Specifically, we ask three questions:
- How do self dsRNAs evade innate immune sensing?
- How do disease associated genetic variants affect dsRNA editing and sensing?
- Which disease relevant cells are the trigger of immune response due to unwanted RNA sensing?
We employ an integrated approach including both computational and experimental tools to address basic questions in dsRNA editing and sensing. By doing so, we aim to bring new insights into autoimmunity and RNA therapeutics.